Laboratory Schedule for 2004, Biology 164
Note: labs may
be rescheduled to respond to experimental difficulties or opportunities.
Labs will be in one of the teaching trailers on Wednesdays, 1:15-5 or possibly longer at times, with 1-3 hours outside of laboratory periods in some weeks.
The purpose of the laboratory of Bio164 is for the class to design experiments, carry them out, generate original data, and make conclusions concerning genetic regulation in yeast. The yeast genome is completely sequenced and around 6000 genes have been identified. They include a number that appear to be ORFs (Open Reading Frames) but may or may not actually encode anything. All yeast ORFs have an ID number that assigns them to a place on a chromosome and the Watson or Crick strand for the sense strand. Most also have a gene name that represents what we know of the function of the gene. Much information about each gene is collected and accessible via the Saccharomyces Genome Database at Stanford. The web site, which you will be accessing frequently for this laboratory, is: http://www.yeastgenome.org/
Microarray analysis enables us to examine the expression of all of these ORFs at once, in response to an environmental or mutational change. We can thus get a view of compensatory regulatory changes that is much more comprehensive than is available in most organisms. We can see if entire pathways change, if many genes involved in one organelle are changed, and many other kinds of integrative change patterns are possible to detect. The class will choose a regulatory question to study using this method. We can get background data and access to relevant literature via the Stanford Microarray Database at: http://genome-www5.stanford.edu/MicroArray/SMD/
The second method we will use is quantitative Reverse Transcription PCR. This method enables one to examine the expression of a single gene in comparison with a standard gene known not to change in expression, in two different RNA samples. The mRNA present is copied into cDNA and a variety of dilutions and replicates enable one to obtain its efficiency of amplification and its amount of amplification compared to our standard gene, TUB1. The real time PCR machine (ABI Prism 7000) will be used with SYBR Green dye and a denaturation analysis following the PCR step for this analysis. Some students may choose to do a second microarray experiment instead of the q RT PCR, but all will analyze the data from both kinds of experiments. The part of the laboratory procedural notes giving procedures for qRT PCR will be handed out later in the semester.
The laboratory performance will be graded
by LH based on her observations of your seriousness of purpose and thinking in
lab (mistakes don't affect your grade unless you seem to be not paying close
attention to detail throughout the semester). Part of this grade
will come from a laboratory notebook that will be graded twice during the
semester, and part from a final report on the microarray experiments that you
will write and hand in with your lab notebook the second time it's submitted for
grading. Use your lab notebook in preparation for and during every
laboratory as a journal of what you will do/are doing. Lab books
should include background material with references for each of the experiments,
including reading and description of both the concepts and the methods used.
They should also reflect everything you have done and when it was done,
and provide enough information to allow someone else to repeat your experiment. You should include the laboratory handouts by pasting them
in if you are using a bound notebook, or by including them as pages if you
are using a loose leaf notebook.
Make sure your lab notes reflect that you have read and assimilated the
laboratory handouts. Also, make sure you have analyzed and commented upon
each result, whether control or experiment in nature.
Sept 1, lab 1. LH
discusses with class the project for the year. Students choose to study an
aspect of DNA repair or an aspect of energy metabolism (the whole class will
study one of these). We will read and discuss papers on each of these and
decide on the topic at this laboratory session. We will also read and
discuss papers on
microarrays and discuss them as a technique (what are they? what
kinds of questions do they help us to address?
what are their limitations or problems?)
Get instructions for logging into the GCAT assessment site and taking the pre
microarray survey there.
Students write up tentative hypotheses in lab notebooks.
Sept 8, lab 2. LH and students discuss the background for DNA repair or energy metabolism, and LH introduces the day’s experiment briefly. Isolation of RNA; see lab handout; add handout to notebook. Be sure to note any deviation from written procedure in your lab notes. We will use total RNA rather than mRNA in our experiment, so we will not isolate polyA+ RNA. Students should continue to write thoughts about hypotheses in lab notebook, using SGD and SMD web sites to look at specific genes/processes that may be affected in their experiment, so that specific genes can be predicted to change or not. If time permits, we will go to SS5 and be introduced to GenePix and GeneSpring software for microarray analysis.
Sept 15, lab 3. LH
calls on two students to review the purpose of the experiment and what was done
last week. Quality control tests on the
RNA prepared. Students will
dilute a tiny sample of their RNA and will also apply another tiny sample of it
to an agarose gel for quality control. During dye coupling, practice
filtering and clustering data using GeneSpring and the
cell cycle data set. Final discussion of background papers on topic of
experiment and microarrays.
Sept 22, lab 4.
LH calls on two students to review the purpose of the experiment and what
has been done so far. LH describes briefly the
Reverse Transcriptase cDNA probe synthesis protocol, cDNA purification, and
hybridization for today.
Synthesis of cDNA and hybridization setup.
During the two hour RNA to cDNA incubation today, we will meet as a class
and discuss further aspects of the project chosen. During this laboratory,
students will practice gridding spots from last year's data using GenePix and
Magic Spot and will practice choosing data for analysis using the cell cycle
data in GeneSpring. Write up
new thoughts about hypotheses about microarray results.
Sept 29, lab 5. LH calls on two students to review the
purpose of the experiment and progress. Students and LH describe and discuss scanning process and the type of data
generated. Class moves to SS5 at
scheduled times (two lab groups can grid at once) and proceeds to grid data with
GenePix. After 1 hour, gridding work must be saved and continued at
another time between laboratories, as a new set of groups will be incoming.
Oct 6, lab 6. Lab
books turned in for grading at the end of the period.
During this lab period we will begin the more sophisticated level of data analysis from the microarrays.
Each group should have
obtained an Excel spreadsheet of the GPR files, imported them into GeneSpring
and filtered the data by the end of the period. This work will continue
next week in laboratory. Between laboratoryies and in these two
laboratories, each group needs to have clustered your data using hierarchical
clustering (treeview) and qCLUSTER methods. You
need to record the results of each clustering in a file, and take notes in your
lab book of apparent functions represented in the genes seen in each of the clusters.
Oct 13, lab 7.
LH will call on two students to summarize the microarray experiment so
far.
Lab books will be returned; continue the data analysis using
GeneSpring and design second array experiments. LH
will note during the grading process what kind of genes might be included in
each cluster, so you can then return to your data files indicated as interesting
by LH and get information on all the genes in them using SGD to get the names
and functions of the gene products, or annotations.
This information needs to be recorded in your lab books, with any
thoughts you have about what each clustering method has done.
In your notebook, comment on how the results compare with your hypotheses
at different points.
Introduction to MAGICTool data analysis program, using the entire class data
set of gene expression ratios. In this laboratory we will design the second
microarray experiment, capitalizing on the results of the first.
Oct 20, lab 8: Begin the second microarray or qPCR laboratory work. Students prepare and present material about the technique of
quantitative Reverse Transcription PCR. The
students using microarrays for a second experiment will plan collection of the
samples for the RNA preparations needed.
Oct 27, lab 9. LH will be away at HHMI program director's meeting Oct 25-Oct 28 so laboratory will be informal and will consist of continuing cluster analyses of your data using GeneSpring and MAGIC Tool at times during the week that are convenient for the student groups. Make sure to note in your lab notebooks when you worked and on what you worked, as well as notes on the results.
Between laboratories: Between laboratories the cells should be grown and harvested and snap frozen for future RNA preparations, according to your approved plans for the next experiment, before the next laboratory period. Also, if doing qPCR, you should redo quality control PCR and/or redesign primers and order new ones if needed; consult with LH and Laty.
Nov 3. lab 10.
Preparation of new RNA samples for arrays or qPCR.
Using the same method as in the
earlier laboratory on RNA preparation, today you will prepare RNA from your
samples of cells you grown and frozen and also t
Nov 10, lab 11. cDNA labeling and hybridization of second set of microarrays or setup of qPCR. We will discuss and review the method before we begin, if possible, one of each lab group doing arrays needs to attend and help with the washing and scanning of the arrays. If doing qPCR, we will collect the data from all experiments in the morning. Two computers in SS5 have the software for the ABI Prism 7000 on them and can be used for studying the data. You should also copy the data from the table at the end of the output into Excel so you can calculate the ratio of expression to our control gene, TUB1. If doing microarrays, you will need to grid your data during this week or next week. Signups for data analysis periods will be scheduled during lab, so bring your calendars!
Nov 17, lab 12. Microarray data sets for each array (Tiff picture files at two different wavelengths) will be put onto CDs and made available for pickup in the laboratory or LH's office. Students need to grid and collect data and give the normalized GPR files to LH for posting in the class data space on the H server by the end of Monday, Nov 22. Sign up for data analysis time the weeks of Thanksgiving and Nov29-Dec3 TODAY in lab; bring calendars!
Nov 24, lab 13. Makeup/data analysis laboratory. If you have fallen behind due to RNA quality control or otherwise need to do wet lab work, this day can be used for it. It's the day before Thanksgiving, so lab is optional. If you've signed up for time to work on data analysis in SS5 during this lab, it will relieve the crunch next week.
Dec 1, lab 14, Data analysis/ work on report on class
project. Using the data
posted for microarrays and the qPCR data collected by others in the class,
complete the data analysis using GeneSpring, MAGICTool, and ABIPrism7000
software plus Excel for the class project.
Dec 8, Last laboratory, wrap up session: students must attend and participate in final discussion; laboratory books and the short paper on the class project due; all materials must be turned in to instructor and all unwanted materials discarded appropriately during this period.
2004 Experiment
GenReg Laboratory Group Project Choices for 2004.
Constraints on everyone:
We will only do ONE group experiment in the class, in order to get what we hope will be a significant amount of data addressing our class question. Each lab group (two students) will be able to do ONE microarray slide in the first round of experiments. In the second round, about half of the lab groups can work on one more microarray and about half can work on testing the expression of genes identified from the microarray experiments by means of real time reverse transcriptase PCR experiments. So, plan your tests/controls so that 6 microarrays, with perhaps 3 more to test vital points or provide replication, will be enough to address your questions/hypotheses.
1. Nucleotide Excision Repair Gene Expression
Background:
Yeast, like most eukaryotic cells, has multiple DNA repair processes. One of the most important in all kinds of cells is nucleotide excision repair, NER. In yeast, a complex of two proteins, Rad1p/Rad10p, is a vital part of the NER pathway that makes incisions near a site of DNA damage such as a thymine dimer or other bulky adduct. Without either of these two proteins, i.e. in strains deleted for either of the two genes encoding the dimer, there is greatly increased sensitivity to UV damage (killing).
1. Paper shows that just a few genes are induced by DNA damage, regardless of the type of damage (but not including UV damage or looking at a mutant affecting NER): Gasch, Audrey P. et al, Genomic Expression Responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol of the Cell 12:2987-3003(2001).
2. Prakash, S and Prakash, L. Nucleotide excision repair in yeast. Mutation Research 451:13-24 (2000). Paper reviews the role of various enzymes in NER process, and shows that complexes exist with the NER proteins associated with other pathway proteins, especially those needed for transcription with RNA polymerase II (TFIIH is a protein that stimulates RNA pol II transcription). Review article providing background and perhaps ideas about what genes might be affected in NER mutants.
Strains available: Wild type W303a; T177, a W303a strain in which rad10 has been deleted and which is therefore deficient in nucleotide excision repair and sensitive to UV irradiation.
Other information and resources:
1. Last year’s class tested T177 compared to wild type without UV treatment and found little or no difference in mRNAs assessed via microarrays; this data set is available to be compared with any new data. Other available data in our lab include microarray data from wild type/wild type comparisons, rad52delete/wild type comparisons (2 available on our computers).
2. We have a Stratalinker which can be used to provide UV irradiation to cells on plates. Cells in liquid don’t receive UV very well, since the water base of the liquid medium absorbs most of the UV energy.
3. UV damage can be repaired by a photoreactivation system that will reverse the damage; in order to prevent photoreactivation from acting to reverse the DNA damage to which you want the cells to respond, you can wrap the container(s) of UV irradiated plates or tubes with aluminum foil to keep out visible light.
4. Attached is a survival curve for wild type and for T177 yeast prepared by Adam Simning for his senior thesis; it may be helpful in designing experiments.
2. Energy metabolism in petite strains from yeast.
Background:
In yeast, if wild type cells are grown on rich medium with ethidium bromide added to it, they tend to lose all or part of their mitochondrial DNA and become respiratory deficient cells that make small colonies (‘petites’, also known as rho minus or r-). This characteristic is inherited by the progeny cells even when propagated without the ethidium bromide medium. Since the mitochondrial genome encodes important subunits for some of the electron transport carriers, the electron transport and oxidative phosphorylation reactions cannot occur; the petite cells must grow by fermentation and can grow without oxygen by fermenting glucose to form ethanol.
Paper 1: Epstein, C. B. et al., Genome-wide responses to mitochondrial dysfunction. Mol Biol of the Cell 12:297-308 (2001). Paper shows microarrays of petites and wild types; of course, every time a new petite strain is isolated, there is a chance that a different part of the mitochondrial genome is deleted so there may be differences between his petites and any we isolate here in terms of their gene expression data. The study suggested an induction of peroxysomes and interaction with the retrograde regulatory genes that participate in mitochondrial/nuclear cross talk in petite strains. A few different inhibitors and media that would allow only anaerobic or only aerobic growth were tested in this paper; others would be interesting. For example, what about adding malonate, a competitive inhibitor of an enzyme of the Krebs Cycle, Succinate DeHydrogenase? What might that do to gene expression in a petite versus wild type?
Paper 2: (Not in packet, just for you to know about!) Taylor, D et al. Conflicting levels of selection in the accumulation of mitochondrial defects in Saccharomyces cerevisiae. Proc Natl Acad Sci, USA 99:3690-3694 (2002). Paper cites human degenerative diseases that result from mitochondrial genomic changes, and analyzes mathematically a situation in which defective mitochondria should come to predominate (within- and among-cell selection values were manipulated by the modeling team). This one is so you will realize the weird ‘petites’ of yeast have potential to help us understand human diseases!
Paper 3: DeRisi, J, Iyer, V and Brown, PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680-686 (1997). Looks at diauxie, which Paper 1 compares with the petite gene expression patterns.
Strains available: W303a wild type, and some strains selected for small colonies on several rounds of ethidium bromide-containing-YPD medium so they presumptively are petites.
Experiment chosen:
Nucleotide Excision Repair in Deletion
Design of Nucleotide Excision Repair Experiment, 2004; concepts prepared by NER team with LH following their guidelines...
Both the W303Ra wild type (YLH208 stock) and the T177 deletion of RAD10 (YLH268 stock) were streaked out on YPD medium and single colonies grown. A well isolated colony was used to inoculate 5 ml YPD liquid medium which was incubated overnight with shaking. At 10 AM, larger culture flasks of YPD were inoculated with 1 ml/100 ml medium from the overnight stock culture. For the wild type, 150 ml was used; for the T177 200 ml was used (since it sometimes grows a bit more slowly). These cultures were grown with shaking until 3:30 PM. Cells were spun out in sterile 50 ml tubes and the media discarded. The cells from each of the two strains were resuspended in about 3 ml PBS and distributed equally into 6 sterile microfuge tubes. The tubes were spun 2.5 minutes at 10K x G and the supernatants discarded. Each was thoroughly resuspended in 100 ul PBS.
For each strain, 3 tubes of cells, meant to serve as the control samples for the three microarrays from that strain, remained at room temperature in the PBS. The remaining three samples were transferred to a YPD agar plate and irradiated with a dose that is predicted to produce 75% viability when photoreactivation is not permitted (where we expect very good repair, as opposed to lots of cell death with heavier doses). For wild type (208) this dose was 25 Joules/meter squared while for the T177 (268) it was 2 Joules/meter squared. Two of the three plates were wrapped with foil immediately after irradiation. All three were incubated at 30 degrees.
One foil wrapped plate’s cells were collected from the agar surface after 5 minutes of recovery, using foil to keep light exposure to a minimum. To collect the cells, 0.7 ml of PBS was micropipetted onto the plate, the surface was swept with a sterile glass spreader, and the suspension was sucked up into a P1000 micropipettor. This procedure was repeated to increase yield of cells. The 5 minute sample and all three controls were then spun out and the supernatant liquid discarded; the cell pellets were frozen at -20 degrees. After 15 minutes at 30 degrees, the cells from the other foil wrapped plate and the plate without foil were resuspended, collected, and frozen as described for the 5 minute plate, keeping light exposure to a minimum for the plate kept under foil during incubation. Therefore, we have the following sets of samples for RNA preparation for use on microarrays.
Control sample: Irradiated sample:
Array 1: Wild type (208) (208) UV, 5 minutes recovery, foil
Array 2: Wild type (208) (208) UV, 15 min recovery, no foil
Array 3: Wild type (208) (208) UV, 15 min recovery, foil
Array 4: T177 (268) (268) UV, 5 min recovery, foil
Array 5: T177 (268) (268) UV, 15 min recovery, no foil
Array 6: T177 (268) (268) UV, 15 min recovery, foil
Protocols provided:
Microarray protocol 1: Preparation of total RNA from budding yeast frozen cell pellets.
Microarray protocol 2: Quality control tests of total RNA of yeast.
Microarray protocol 3: ISB method for direct labeling of cDNA with CyDyes.
Microarray protocol 4: Hybridization and washing of arrays.
Microarray protocol 5: Scanning, gridding, and intensity collection from microarrays with GenePix.
Microarray protocol 6: Analyzing data with GeneSpring and MagicTool.
Microarrays, Protocol 1:
Preparation of total RNA from yeast with Qiagen RNeasy kit. (Notes based on Michelle Wu protocol/LH lab)
Qiagen RNeasy Mini kit #74104
Qiagen DNase I kit #79254
Sigma Acid washed glass beads #G-8772
Sigma b-mercapto-ethanol #M6250
Special notes on procedures:
Use a maximum of 2.5´108 cells per column.
The glass beads may stick to the outside of the screw cap tube along the ridges, preventing a proper seal of the tubes during breaking of the cells. Try NOT to get beads on this area. If you do, consult the instructor.
Add 10 ml b-mercapto-ethanol per 1 ml RLT buffer in the kit (stable for 1 month after addition of b-ME).
After disruption, all steps of the protocol should be performed at room temperature. Work quickly through the procedures. Also do not let the centrifuge cool below 20ºC.
Each aliquot of DNase I is 21 ml and stored in the microarray box in the -20ºC freezer. Add 140 ml buffer RDD, stored at 4 ºC, for 2 sample digestion.
USE RNA PARANOIA THROUGHOUT PROCEDURE!! Gloves, RNase Erase all benches, do not open tubes with bare hands, try to only handle tubes through gloves to keep RNase fingerprints as far away as possible.
Glass bead grinding of cells.
DNase Digestion
14. Add 140 ml buffer RDD to the 21 ml aliquot of DNase I. Gently pipet to mix. DNase is especially sensitive to physical denaturation, mix gently, do NOT vortex.
Total RNA absorbance measurements.
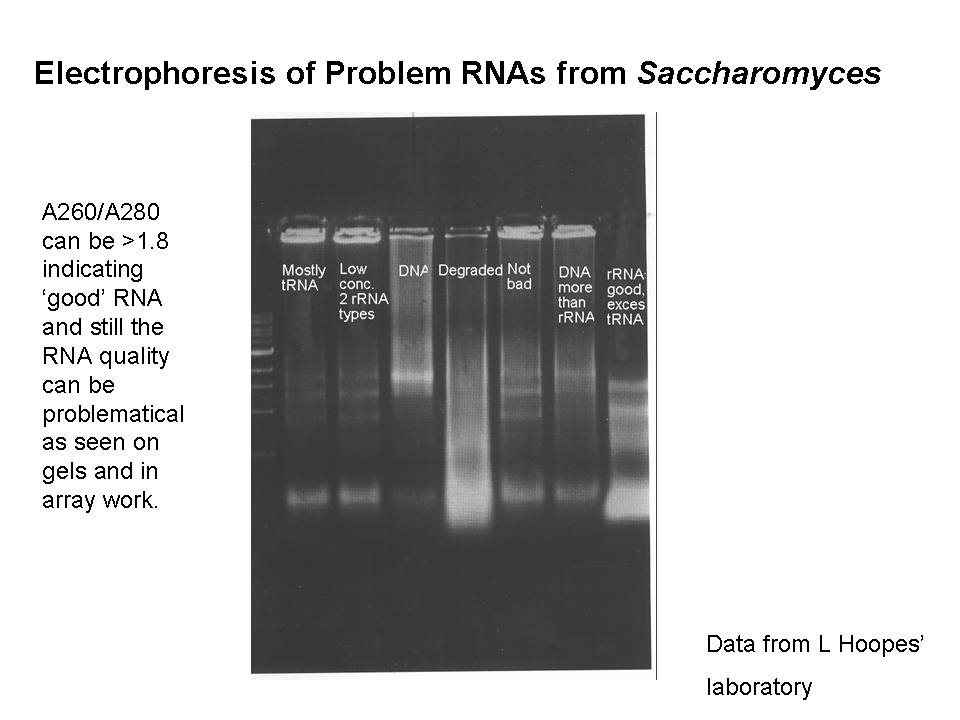
Microarrays, Protocol 3:
Direct Incorporation of Cy3/Cy5 During Reverse Transcription(Method from Institute for Systems Biology, 2003, with notes by Todd Eckdahl and Anne Rosenwald)
Materials Needed:
Microarray slides (70-mer plus-strand oligomers)
RNase-free water
oligo dT primer (16- to 18-mer) at 1 mg/ul
Coverslips, 22 x 40mm size from Corning
100 mM DTT (dithiothreitol)
low dTTP dNTP mix (10 mM each dATP, DCTP, dGTP, 1 mM dTTP)
Cy3-dUTP and Cy5-dUTP (1 mM each [separately])
3 M Ammonium Acetate, pH 5.2 (OR 3M Sodium Acetate, pH5.2)
100% Ethanol, 70% Ethanol
Enzymes:
Superscript II Reverse Transcriptase, 5X first strand buffer
RNase A (4 mg/ml)
RNase H (2 unit/ml)
Degrade RNA
Purification (Using Qiagen PCR CleanUp Kit)
Microarray Protocol 4: Hybridization and Washing of Microarrays.
(Based on notes of Todd Eckdahl from procedures of Institute for Systems Biology, 8/03)
Microarray Slide Processing
This procedure is optional for oligo slides (Isb or WU slides); it redistributes the oligo DNA on the slides, which helps spot morphology and hybridization. It is mandatory for PCR product slides (TMC or Stanford slides) , otherwise the duplex DNA will not be opened up for hybridization. Some PCR product slides may not have had earlier steps in post printing processing done and may need additional steps; check with the array providers to be sure.
1. Steam the DNA side of the slide over boiling dH2O. Do not allow visible droplets to form on the slide.
2. Immediately place the slide (DNA side up ) on a heat block or hot plate set to 100 C or slightly less to snap dry. Take off after 5 seconds.
3. Repeat steam step, followed by drying step. Allow the slide to sit on the heat block for 15 seconds this time. Allow slide to cool.
Prehybridization and Blocking
1. Place slide into a 50 ml tube filled with warm (55 C) 3x SSC, 0.1% SDS, 0.1 mg/ml Sonicated Salmon DNA.
The slide must be completely immersed.
2. Agitate gently for 30-60 minutes at room temp. by rocking on a platform (lay the tube down flat on the rocker but
wedge it so it will not roll off).
3. Quickly transfer the slide to a 50 ml tube with dH2O. Dip several times.
4. Blow the slide dry with air or spin it 5 mintues in a clinical centrifuge in a dry 50 ml tube. If blowing dry, the idea is to chase all the drops of water off the slide while it is held at an angle on a towel. If drops of water start to dry in place on the array, quickly immerse the slide back into water and start again. You are not trying to blow dry the slide, rather you are trying to push the liquid away from the spots. If you see streaks at this stage, rewet the slide. If you see dried-on streaks, you will have streaks in your final scan.
Labeled Sample Preparation and Hybridization
1. Combine 30 ul (entire sample) of each cDNA with its dye into a single tube and Speed-vac to 1-2 ul. If
sample dries (avoid this if possible!!), add 2 ul DEPC-H2O and let stand 2 minutes (slight warming is OK).
Posthybridization Washing
1. Heat 50 ml 1X SSC / 0.1% SDS and 0.5X SSC to 55 C.
Microarray Protocol 5: Scanning, gridding, and data collection
Scanning on AxonGenepix scanner.
We use our Axon GenePix 4000B scanner, currently located in SS5, to scan the microarrays. For more information about the material below, you can refer to the help tab in the GenePix Pro software for more about any item given in italics.
X axis: Fullscale
Y axis: Log Axis on Fullscale
The histogram that appears shows you the percentage of Normalized Counts that are at a given Intensity. Note that the histogram only shows you the pixels that you are viewing in the image tab; i.e., if you are zoomed in on the image, it will only show the zoomed in area in the histogram. Remember that every pixel is represented in the histogram, so artifacts and dirt will skew the readings. If you have a lot of artifacts or dirt, try to zoom in on a clean portion of the array to determine more appropriate PMT settings. You can avoid the dirt later in the gridding process. Adjust the two PMTs during this ongoing data scan so that you can see similar histograms of pixels across the entire intensity range. Note, though, that saturated pixels (with counts greater than ~67,000) will be thrown out and spots with pixel counts close to background will result in poor data, so you don’t want to ‘correct out’ all of your intensity in either channel.
GenePix 5.0 Spot Finding
Preparing the image for quantitation:
1. Open GenePix 5.0 with the dongle installed in the computer.
2. Click on the right side menu on the disc to bring up stored files; choose Open Image, then find the file you are using and select the two TIFF files (green and red) that need to be opened. (You will need to hold down the shift key when selecting the second one).
3. Load the grid file: click on the disc icon on the right and choose Open Setting. Then find the file you need with the gps settings for the chips we are currently using. Double click on the desired file to load.
4. Set the left side menu to “block mode” (square with up arrow on left side). Now, click on the Zoom key (magnifying glass). Then you can use click at upper left and drag to desired lower right to select the part of the image with your data and the grids in it and zoom it to full screen.
Preliminary positioning of grids for automatic spot finding:
Refine positions of spots before collecting data.
Data capture: collecting a table of intensities in a 'gpr' (gene pix results) file; deriving a working copy in an Excel file.