|
HOME | DESIGN | MICROARRAY SAMPLES | NORMALIZATION | SIGNIFICANCE TESTING | SAM | H. CLUSTERING | PAM CLUSTERING | PAM CLASSIFICATION | CONCLUSIONS |

This web page was produced as an assignment for a course on Statistical
Analysis of Microarray Data at
Hierarchical Clustering:
To examine the effectiveness of hierarchical clustering, we performed
clustering on two subset of miRNAs - 20 random miRNAs, and 20 significant ones.
Random miRNAs:
R was used to select 20 random indices from our total, merged miRNA data (768 miRNAs possible).
Presumably, there is no relationship between these miRNAs,
so that any clustering observed is just a remnant of the fact that hierarchical
clustering always must create clusters, even when they are not
significant.
Below are dendrograms from the clustering.
Below each dendrogram is a brief explanation of
both the distance method and clustering method used. Note that the
heights of the trees differ because they are a direct reflection of the linkage
and distance methods used. So, for example, because 1-correlation yields
a maximum distance of 2, we would not expect the trees to get any higher than
2. Likewise, complete linkage (which measures cluster distances based on
the farthest elements) will yield much higher trees than average linkage (which
measures cluster distances based on the average of all the elements).
|
|
|
|
|
|
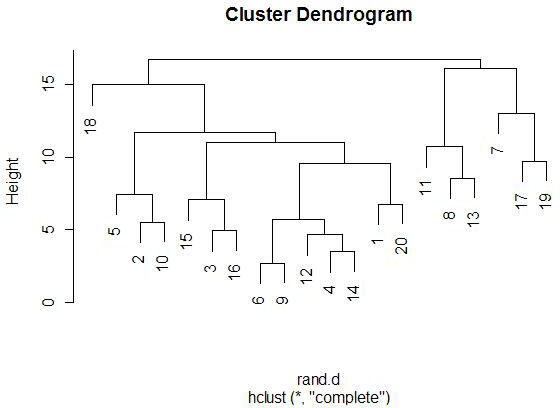
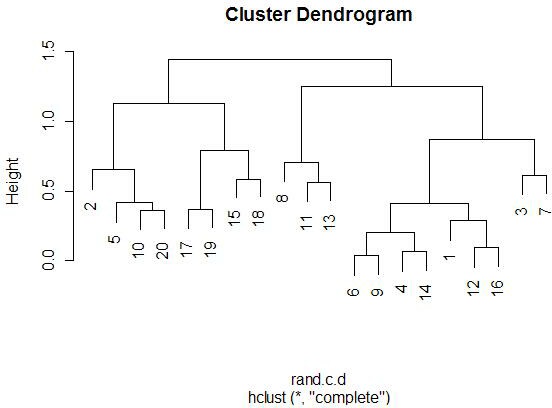
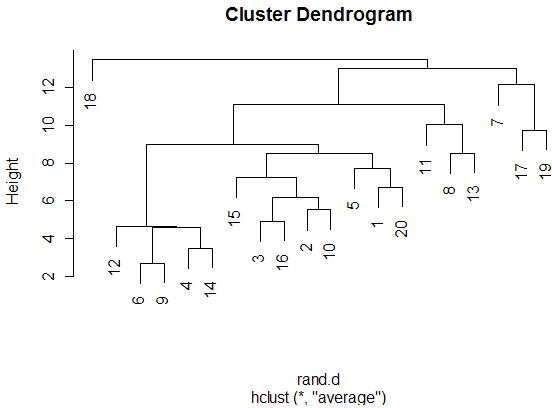
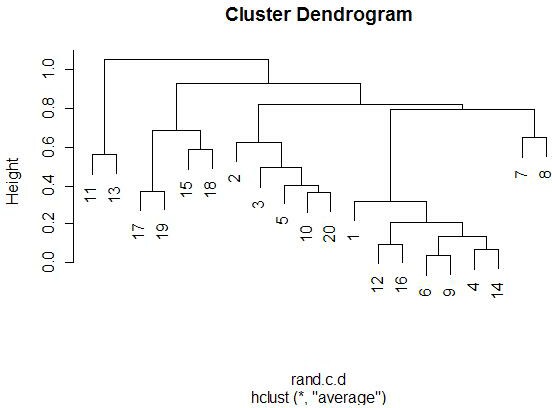
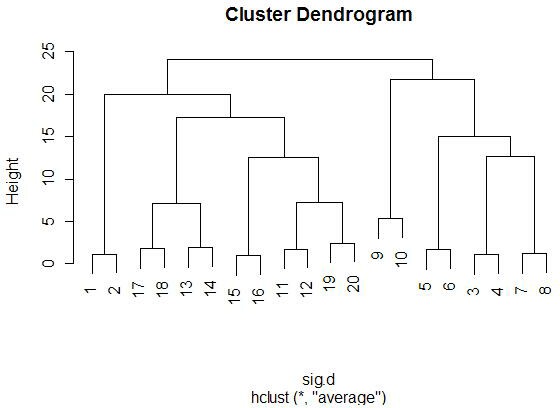
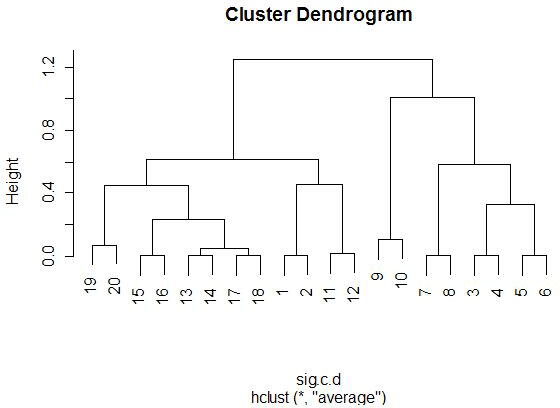
With the graphics above, it is meaningful to compare the graphs that are both
side-by-side, as well as top-and-bottom. Side-by-side trees use the same
linkage method, and top-and-bottom trees use the same distance method.
Interestingly, the euclidean distance trees clustered
with average and farthest methods both retain some of the same clusters
(7-17-19, 11-13-8). Similarly, for the 1-correlation distance trees, the
clusters are all somewhat similar, especially pair-wise clusterings
(17-19, 15-18, 12-16). This makes sense because the farthest element
heavily influences the average calculation (since the average is not robust).
So, these results make sense).
Comparing the same linkage methods with different distance methods
(side-by-side comparisons) yields less similarity between the clusters.
The general location of the different miRNAs is
somewhat retained, but we see less similar clusters and less similar pairings.
When comparing caddy-corner from one another, we again see a decrease in
similarities. One notable exception is the 8-11-13 cluster, which appears
in three of the four trees. These miRNAs must
be very similarly expressed in both magnitude (Euclidean measure) and trend
(correlation measure). We also see 17-19 paired together in every tree,
with the same interpretation.
Significant miRNAs:
Because the previous analyses were done with the uncompressed data (that
is, the spots were not merged), I decided to do this analysis with the
uncompressed data as well. It actually affords an interesting control,
because spots that are the exact same miRNA
should cluster together no matter what.
Below is a table of the significant genes used:
|
Spot Number |
miRNA name |
|
1146 |
hsa_miR_122a |
|
1122 |
hsa_miR_122a |
|
993 |
hsa_miR_145 |
|
969 |
hsa_miR_145 |
|
75 |
hsa_miR_29b |
|
51 |
hsa_miR_29b |
|
523 |
ambi_miR_7510 |
|
499 |
ambi_miR_7510 |
|
197 |
hsa_miR_214 |
|
221 |
hsa_miR_214 |
|
1421 |
hsa_miR_422b |
|
1397 |
hsa_miR_422b |
|
1499 |
hsa_miR_1 |
|
1523 |
hsa_miR_1 |
|
695 |
(blank) |
|
719 |
(blank) |
|
1419 |
hsa_miR_133a |
|
1395 |
hsa_miR_133a |
|
1448 |
hsa_miR_422a |
|
1472 |
hsa_miR_422a |
Below are dendrograms from the clustering.
Below each dendrogram is a brief explanation of
both the distance method and clustering method used.
|
|
|
|
|
|
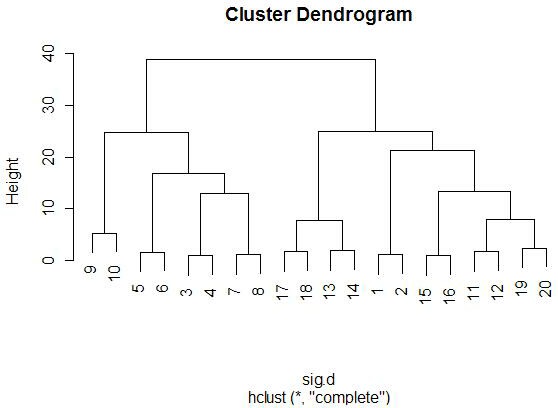
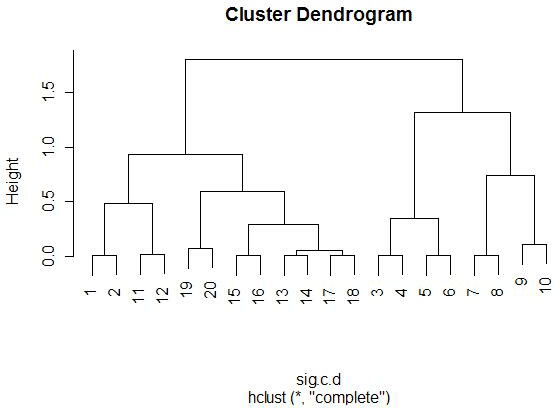
There are a few things to note from these diagrams. First, happily, all
of the neighbors (1-2, 3-4, 5-6, etc.) represent the exact same miRNA, so it's a good thing that they are all clustering
together first.
When we take a step back and look at the larger clusters (realizing that the pairwise ones can be considered one group), we see some
similarities between all of the clustering techniques. The cluster
(3-4-5-6-7-8-9-10) appears in all four trees as a highly isolated branch.
In fact, this can be taken one step further to say that each tree divides
into two main clusters - the one just described and then the left-overs.
When looking at smaller clusters, we see more similarities between all four
trees. One obvious one is the cluster (13-14-17-18), which appears in all
four trees as closely linked. Several times, there are clusters that
contain the same elements but are just paired up in a different order, such as
(3-4-5-6-7-8-9-10). Interestingly, in this case, (3-4) links first to
(7-8) when using Euclidean distance, but when using 1-correlation distance,
(3-4) links first to (5-6).
Comparing these results to the random miRNA results
shows that these results are much more consistent across different distance and
linkage methods. One might go so far as to say that one should use
multiple clustering methods before determining that a cluster is significant.
Because hierarchical clustering always results in a tree, it seems like a
good way to check if your tree is significant is to calculate it multiple ways and
see if it remains the same. In our case, anyway, we would quickly be able
to determine which trees are from the random miRNAs
and which are from the significant ones.